May 1531, 15018
Authors
Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell APW, Fisher C, Suciu M, Martyn GE, Norton LJ, Zhu C, Kurita R, Nakamura Y, Xu J, Higgs DR, Crossley M, Bauer DE, Orkin SH, Kharchenko PV, Maeda T
Science 2016, doi:10.1126/science.aad3312.
Abstract
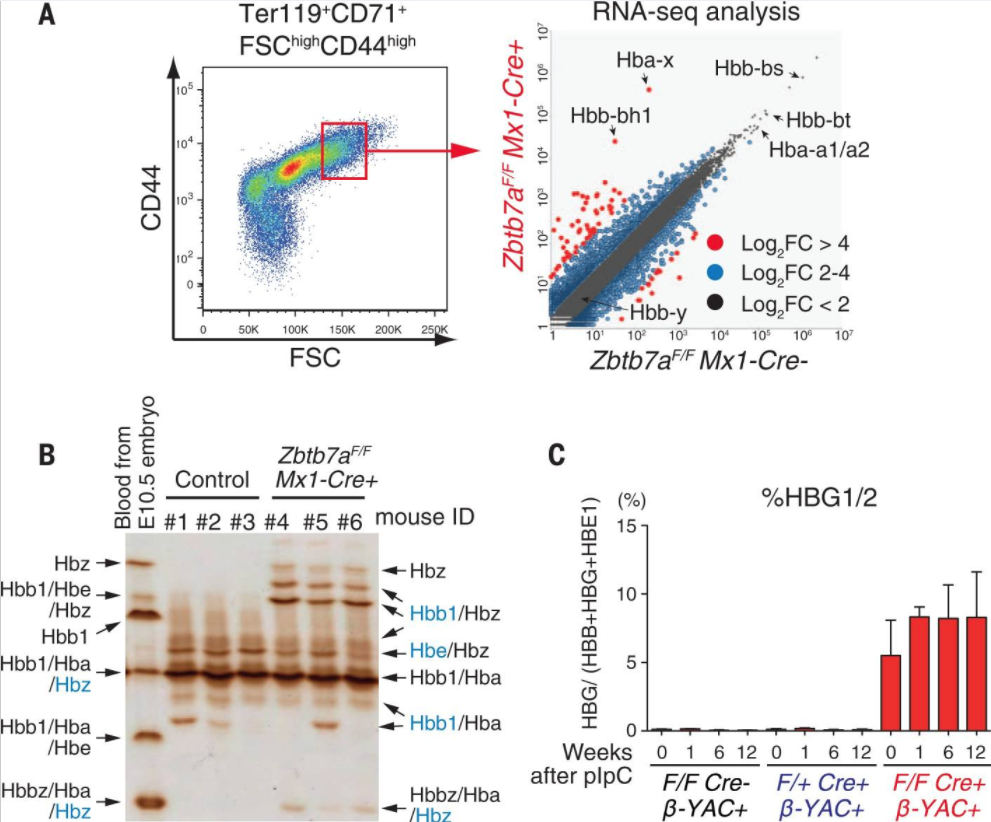
Genes encoding human β-type globin undergo a developmental switch from embryonic to fetal to adult-type expression. Mutations in the adult form cause inherited hemoglobinopathies or globin disorders, including sickle cell disease and thalassemia. Some experimental results have suggested that these diseases could be treated by induction of fetal-type hemoglobin (HbF).
However, the mechanisms that repress HbF in adults remain unclear. We found that the LRF/ZBTB7A transcription factor occupies fetal γ-globin genes and maintains the nucleosome density necessary for γ-globin gene silencing in adults, and that LRF confers its repressive activity through a NuRD repressor complex independent of the fetal globin repressor BCL11A. Our study may provide additional opportunities for therapeutic targeting in the treatment of hemoglobinopathies.